
基因转录调控顺式作用元件包括核心启动子和增强子;核心启动子在转录起始位点(TSS)上游25-30bp位置,比较保守;增强子序列能够招募转录因子,控制核心启动子的转录活性,在基因转录调控中具有重要的地位。在植物中,顺式作用元件,尤其是增强子,在基因组上的分布是不清楚的。ATAC-seq作为获取染色质开放区域强有力的新技术,能够在全基因组水平上研究植物的整体调控区域;相对于传统DNase-seq技术,ATAC-seq技术使用的细胞核数目少(最多5万个),操作简单,因此被广泛采用。
埃默里大学的Roger B. Deal课题组在Plant Cell杂志上发表植物ATAC-seq里程碑式的文章,开启了ATAC-seq技术在植物研究领域的应用。文中作者使用ATAC-seq技术系统研究拟南芥、水稻、苜蓿、番茄的根尖组织染色质开发区域,发现开放染色质区域位于转录起始位点上游3kb内,并且鉴定到4个关键转录因子在根尖中调控一系列保守基因的表达;最后作者比较了拟南芥根毛细胞和非根毛表皮细胞染色质开放区域及根毛关键转录因子调控网络。下面小编将详细对这篇文章进行讲解。

ATAC-seq实验设计
- 拟南芥根尖组织ATAC-seq,使用链霉亲和素磁珠富集细胞核;2生物学重复;命名为:INTACT-ATAC-seq;
- 拟南芥根尖组织ATAC-seq,蔗糖梯度密度离心富集细胞核;2生物学重复;命名为:Crude-ATAC-seq;
- 拟南芥根尖组织DNase-seq,蔗糖梯度密度离心富集细胞核;2生物学重复;命名为:DNase-seq;
- 苜蓿根尖组织ATAC-seq,使用链霉亲和素磁珠富集细胞核;2生物学重复;
- 番茄根尖组织ATAC-seq,使用链霉亲和素磁珠富集细胞核;2生物学重复;
- 水稻根尖组织ATAC-seq,使用链霉亲和素磁珠富集细胞核;2生物学重复;
- 拟南芥根毛细胞ATAC-seq,使用链霉亲和素磁珠富集细胞核;2生物学重复;
- 拟南芥非根毛表皮细胞ATAC-seq,使用链霉亲和素磁珠富集细胞核;2生物学重复;
注:所有植物株系都为表达生物素连接酶 - NTF(定位于细胞核核膜)融合蛋白的转基因材料,因此可以使用链霉亲和素磁珠富集细胞核。
生信分析结果解读
1. INTACT-ATAC-seq、Crude-ATAC-seq和DNase-seq三种方法的比较
首先作者对NTACT-ATAC-seq和Crude-ATAC-seq结果进行比较,发现两者鉴定的染色质开放区域结果类似,差别在于NTACT富集的细胞核更纯,线粒体和叶绿体基因组污染更少,数据利用率更高;
接着对INTACT-ATAC-seq和DNase-seq结果进行比较, INTACT-ATAC-seq鉴定到23288个开放的区域(THSs),绝大部分THSs(19516/23288)在DNase-seq中富集;说明ATAC-seq和DNase-seq都能够真实反应染色质开放区域;
三者在基因组上开放区域一致:约75%的THSs都位于TSS上游2k和TTS下游1kb区域。
小结:Crude-ATAC-seq、INTACT-ATAC-seq与DNase-seq在获取染色质开放状态性能上效果相当;Crude-ATAC-seq虽然具有大量线粒体和叶绿体基因组残留,但不需要转基因操作,而且对任何材料适用,因此工作效率更高。
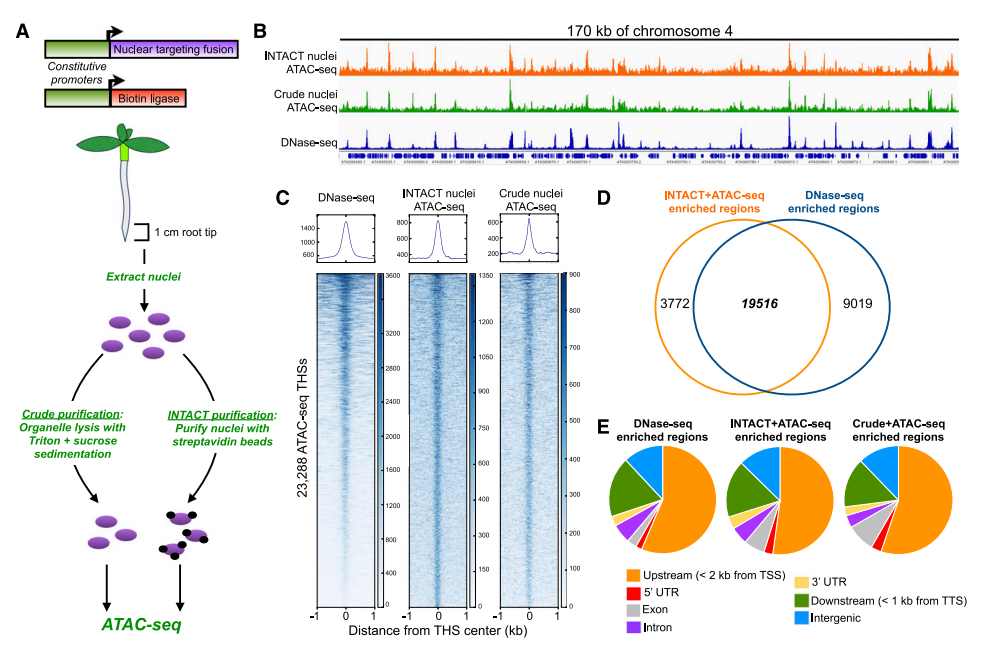
图1:ATAC-seq与DNase-seq结果比较
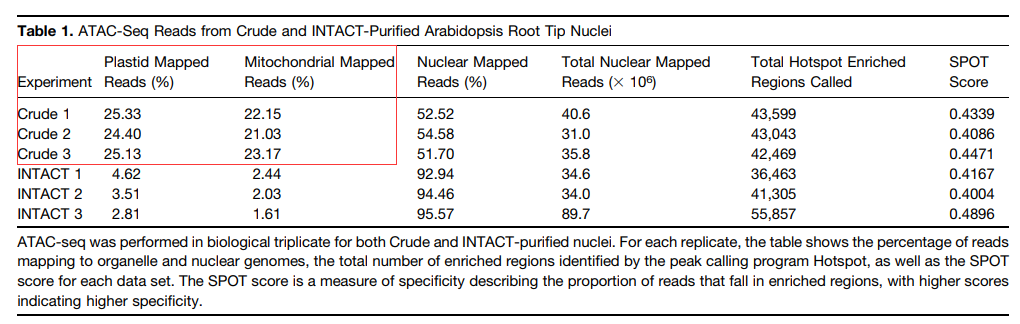
图2:Crude ATAC-seq和INTACT-ATAC-seq线粒体和叶绿体DNA残留率比较
2. 拟南芥、水稻、苜蓿、番茄根尖组织ATAC-seq结果比较
作者首先在四个物种基因组上发现一段相互对应的染色质区域(syntenic region),在此区域上四个物种具有相似的染色质开放模式。
接着对四个物种的THSs分析发现:约70—80% THSs分布在翻译区域以外,按比例排序依次为:上游近端(TSS上游2kb以内范围)、下游近端(TTS下游1kb范围内)和基因间区(TSS上游2kb以外和TTS下游1kb以外)。四个物种大部分THSs位于TSS上游3kb以内,暗示大部分顺式调控元件位于核心启动子近端。
对THSs进一步分析发现,THSs位于上游近端和下游近端比例与基因组大小负相关,基因组越小该区域比例越大:拟南芥基因组最小,只有120Mb,上游近端比例为52%;水稻基因组约400Mb,上游近端比例为37%;苜蓿基因组约480Mb,近端上游比例为30%;番茄基因组820Mb,近端上游比例为11%。下游近端也存在相同的变化模式:拟南芥17%、水稻和苜蓿12%、番茄6%。
而THSs位于间区比例与基因组大小正相关:拟南芥12%、水稻和苜蓿30%、番茄50%。
从THSs与基因关联层面分析发现:拟南芥、水稻和苜蓿, 70%的基因只有一个THSs与其相关联,20%的基因与两个THSs相关联;而番茄正好相反,只有一个THSs位点的基因只占27%,大部分基因具有多个THSs,暗示番茄基因具有较多的顺式作用元件。
小结:四个物种THSs在基因组上具有相同的分布模式;大部分THSs在翻译区域外,并且集中在TSS上游3kb内的范围;拟南芥、水稻和苜蓿大部分基因只有一个THSs位点,而番茄基因具有多个THSs位点,暗示番茄调控元件的复杂性。
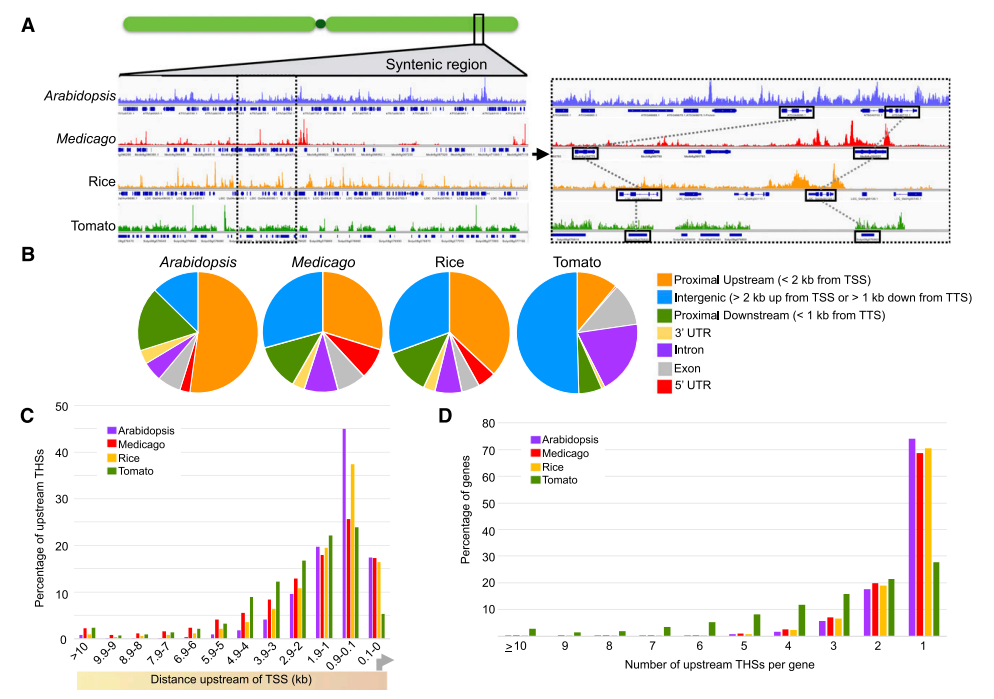
图3:拟南芥、水稻、苜蓿、番茄染色质开放区域比较
3. 拟南芥、水稻、苜蓿、番茄同源基因THSs的特征比较
作者首先在四个物种中鉴定到了373个同源基因,并且在基因TSS上游5kb范围类统计THSs的数量,发现同源基因上游的THSs数量没有保守性,只有少数同源基因具有相同的THSs数量。番茄中,这373个基因至少有一个THSs;而在拟南芥、水稻和苜蓿中,大部分同源基因在TSS上游5kb内未发现THSs。而且同源基因上游开放区域位置分布在四个物种中没有一致性。
接着作者分析了373个同源基因THSs分布区域与基因表达情况的关联性。发现上游区域THSs数目与基因表达量没有关系;暗示THSs不只是简单的激活元件,也是转录抑制因子的结合位点,这进一步导致基因转录调控的复杂性。
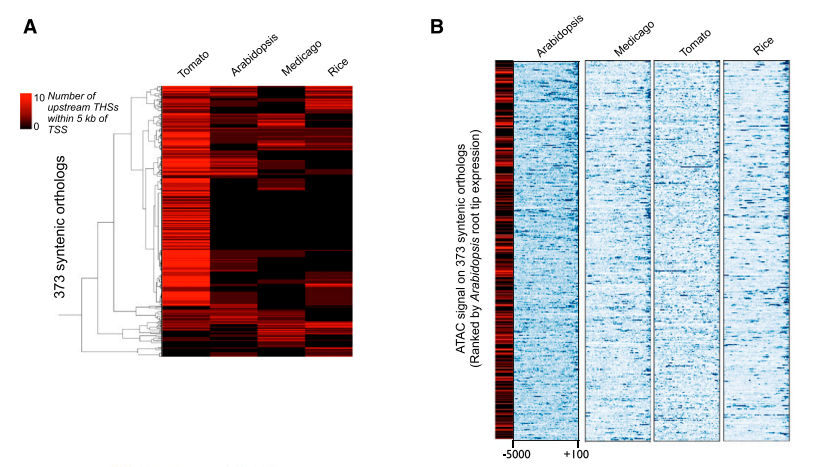
图4:拟南芥、水稻、苜蓿、番茄同源基因相关的THSs数目和位置分析
4. 拟南芥、水稻、苜蓿、番茄转录因子调控特征分析
作者首选对THSs进行TF motif分析,发现30个TF motif在4个物种中普遍存在,并挑选其中4个有价值的TFs进行分析:HY5、ABF3、CBF2和MYB77。
接着使用拟南芥ATAC-seq数据查找含有上述4种TFs motif的THSs,预测TFs的结合位点;使用4种TFs已经发表的ChIP-seq或者DAP-seq数据,分析ATAC-seq预测的结合位点与实际的结合位点是否具有一致性?结果发现:拟南芥中1316个THSs存在ABF3 motif,其中1279个(97%)THSs与ABF3 的ChIP-seq peak位置重合;89%的CBF2预测结合位点与其DAP-seq peak位点重合;74%的MYB77预测结合位点与其DAP-seq peak位点重合;61%的HY5预测结合位点与其DAP-seq peak位点重合;
作者最后分析了包含至少两种转录因子结合位点的基因;发现这四种转录因子具有广泛的共同结合调控的基因,例如,ABF3结合的1271个基因中有297个基因(23%)也能被HY5结合;这297个基因中,有46个被ABF3结合;并且其中有7个基因被4个转录因子同时结合;
由于水稻、苜蓿和番茄中缺乏这4种转录因子的ChIP-seq或DAP-seq数据,作者使用这四个转录因子在拟南芥中的motif进行分析,首先获得含有对应TFs motif的THSs,接着分析THSs对应的基因,最后统计转录因子共同结合的基因比例,总共把结合的基因类型分成16个模块(与拟南芥的分析类似),对于每一个模块调控的基因比例,四个物种较一致。
小结:作者鉴定到了HY5、ABF3、CBF2和MYB77四个转录因子,发现它们在根尖发育过程中具有重要的调控作用,并且结合在TSS上游5kb的范围内,共同结合一系列基因表达。
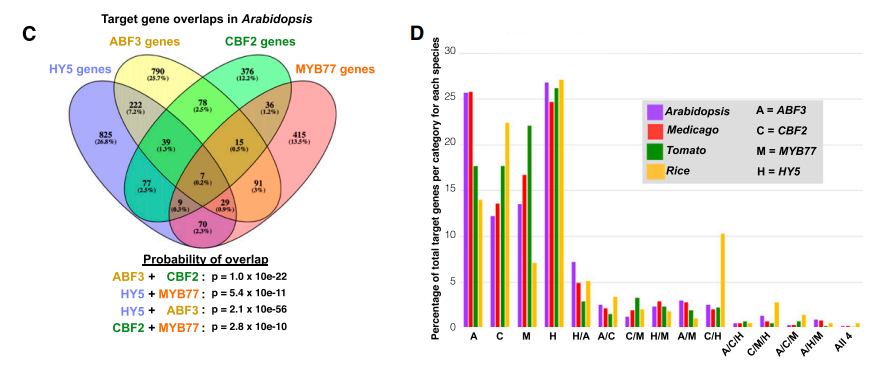
图5:根尖组织中四种关键转录因子共同结合的基因分析
5.拟南芥根尖、根毛细胞和非根毛表皮细胞染色质开放区域比较分析
分析完四个物种根尖组织染色质开放区域的异同及四种关键转录因子的调控特征后;作者进一步比较了拟南芥根尖、根毛细胞和非根毛表皮细胞染色质开放区域。主要结果如下:
首先作者对根尖组织、根毛细胞和非根毛表皮细胞的ATAC-seq数据进行可视化展示,发现三者reads在基因组上分布整体上高度一致;
进一步统计THSs的特征发现,根尖组织存在32942个THSs,根毛细胞存在35552个THSs,非根毛表皮细胞有28912个THSs;其中绝大部分的THSs是三者共有的(总共18742个);6562个THSs只在根毛细胞和非根毛表皮细胞中存在,暗示这些THSs为根部表皮细胞特异的调控区域;
对三种材料特异的THSs统计发现:根尖组织中存在10455个特异的THSs,根毛细胞有7537个特异的THSs,非根毛表皮细胞有2574个特异的THSs;根毛和非根毛各自特异的THSs(命名为dTHSs)可能是导致这两种不同细胞类型形成的重要因素。
作者使用根毛和非根毛表皮细胞归一化的数据进行dTHSs区域的热图展示,可以明显看出这些区域在这两种材料中的开放性显著不同;接着对这些dTHSs进行关联基因分析,发现根毛细胞7537个特异的THSs关联到6008个基因;非根毛表皮细胞2574个特异的THSs关联到2295个基因,基本上一个THSs关联一个基因;
最后作者使用已发表的拟南芥根毛和非根毛RNA-seq数据,发现有3282个基因在根毛中上调表达,其中743个基因能够与根毛dTHSs相关联;258个基因与非根毛表皮dTHSs相关联;108个基因与两种细胞类型dTHSs相关联;2731个基因在非根毛表皮细胞中上调表达,其中156个基因与非根毛表皮细胞dTHSs关联;516个基因与根毛dTHSs相关联;52个基因与两种细胞类型dTHSs相关联;表明细胞中上调表达的基因优先与其特异的dTHSs相关联,但也存在高表达的基因与非开放的区域相关联,符合基因表达先激活、后抑制的规律。
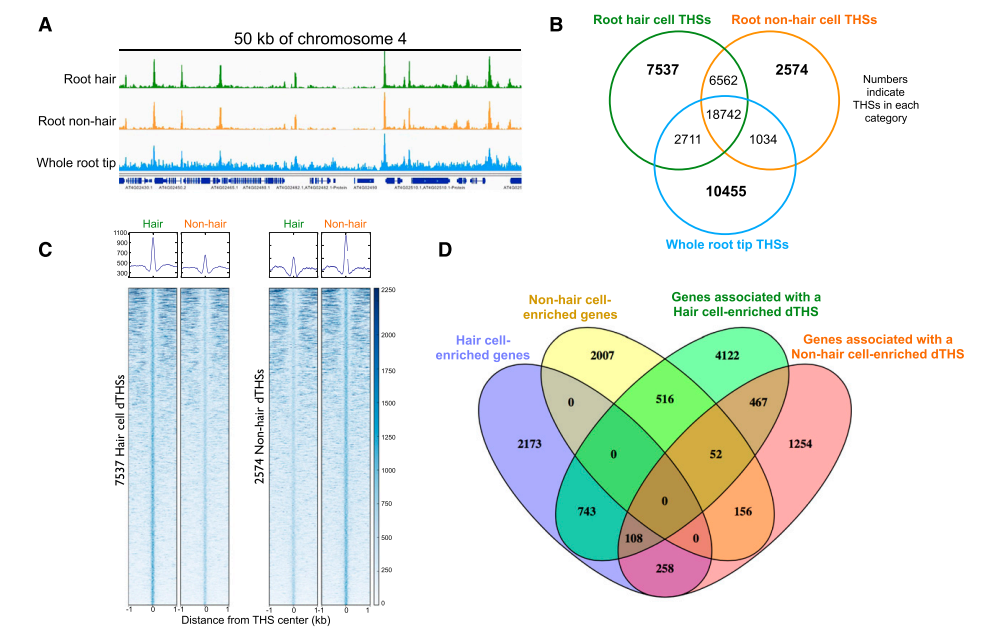
图6:拟南芥根尖组织、根毛细胞、非根毛表皮细胞染色质开放区域比较
6. 根毛细胞和非根毛表皮细胞转录因子调控分析
转录因子对根毛细胞和非根毛细胞命运分化具有重要的调控作用;作者使用dTHSs获得两种细胞特异的motif,并关联到对应的TFs;进一步通过TFs的表达量,获得细胞特异的关键转录因子:根毛细胞中鉴定到4个关键转录因子(MYB33、ABI5、NAC083、At5g04390);非根毛细胞中鉴定到1个关键转录因子WRKY27;
与上文TFs的分析思路一样:筛选含有转录因子motif的THSs,结合TFs的ChIP-seq或者DAP-seq peak数据,筛选与peak重合的THSs,作为最终TFs的结合位点;其关联的基因作为TFs的靶基因,并且与根毛细胞和非根毛细胞各自特异上调表达的基因进行关联分析(绘制韦恩图);
作者首先统计了MYB33、ABI5、NAC083三个转录因子分析的结果:17-21%结合基因在根毛细胞中上调表达;6-9%结合基因在非根毛上皮细胞上调表达;暗示大部分结合基因没有出现差异表达。
接着作者研究了MYB33、ABI5、NAC083共同结合的基因,发现:ABI5和NAC083共同结合207个基因;ABI5和MYB33共同结合238个基因;三者共同结合50个基因; ABI5和MYB33共同结合的238个基因中,有57个基因在根毛细胞中上调表达。作者使用GO富集分析研究这些基因的富集功能;发现ABI5/MYB33结合的238基因显著富集到“响应ABA、水、盐离子、冷刺激”;其中57个在根毛细胞中上调表达的基因,有7个基因被注释到“转录调控”。暗示ABI5和MYB33可能在根毛细胞中处于转录调控级联的顶端。
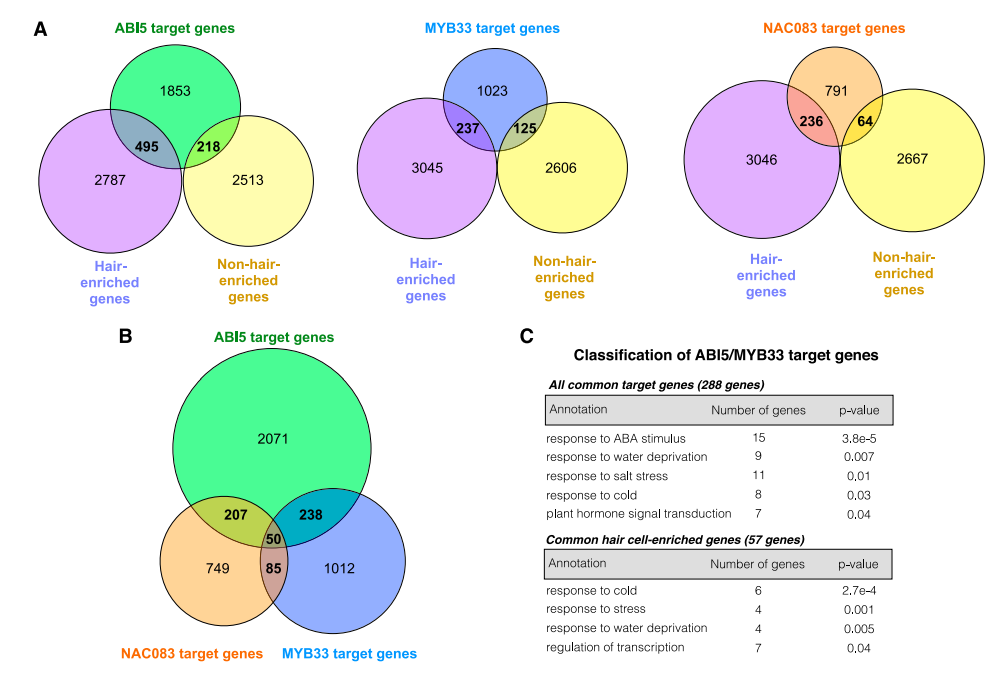
图7:根毛细胞关键TFs调控的基因
总结
纵观全文,可以归结为三个层面:
- 植物根尖组织ATAC-seq技术的开发,发现通过密度梯度离心富集细胞核的方法能够真实反应材料染色质情况。
- 四种植物根尖组织染色质开放区域的比较和关键转录因子调控的分析;
- 根毛细胞和非根毛细胞染色质开放区域的比较和关键转录因子调控的分析;
康测科技植物ATAC-seq技术效果展示
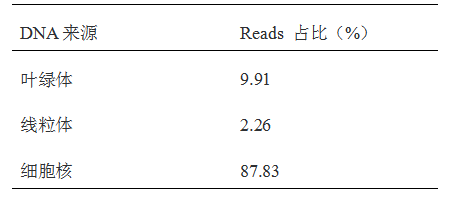
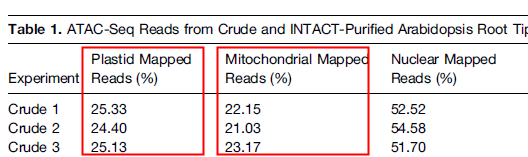
1. 康测科技采用优化的密度梯度离心技术提取细胞核,不需要进行生物素连接酶 - NTF(定位于细胞核核膜)融合蛋白转基因操作,也能获得较纯的细胞核;把线粒体和叶绿体DNA残留量最大限度的降低:线粒体残留2.26%、叶绿体残留9.91% !极大增加了数据的使用效率!
康测科技植物ATAC-seq叶绿体和线粒体残留统计
文献密度梯度离心法叶绿体和线粒体残留统计
2. 拟南芥叶片组织ATAC-seq染色质开放区域统计和转录因子靶基因预测
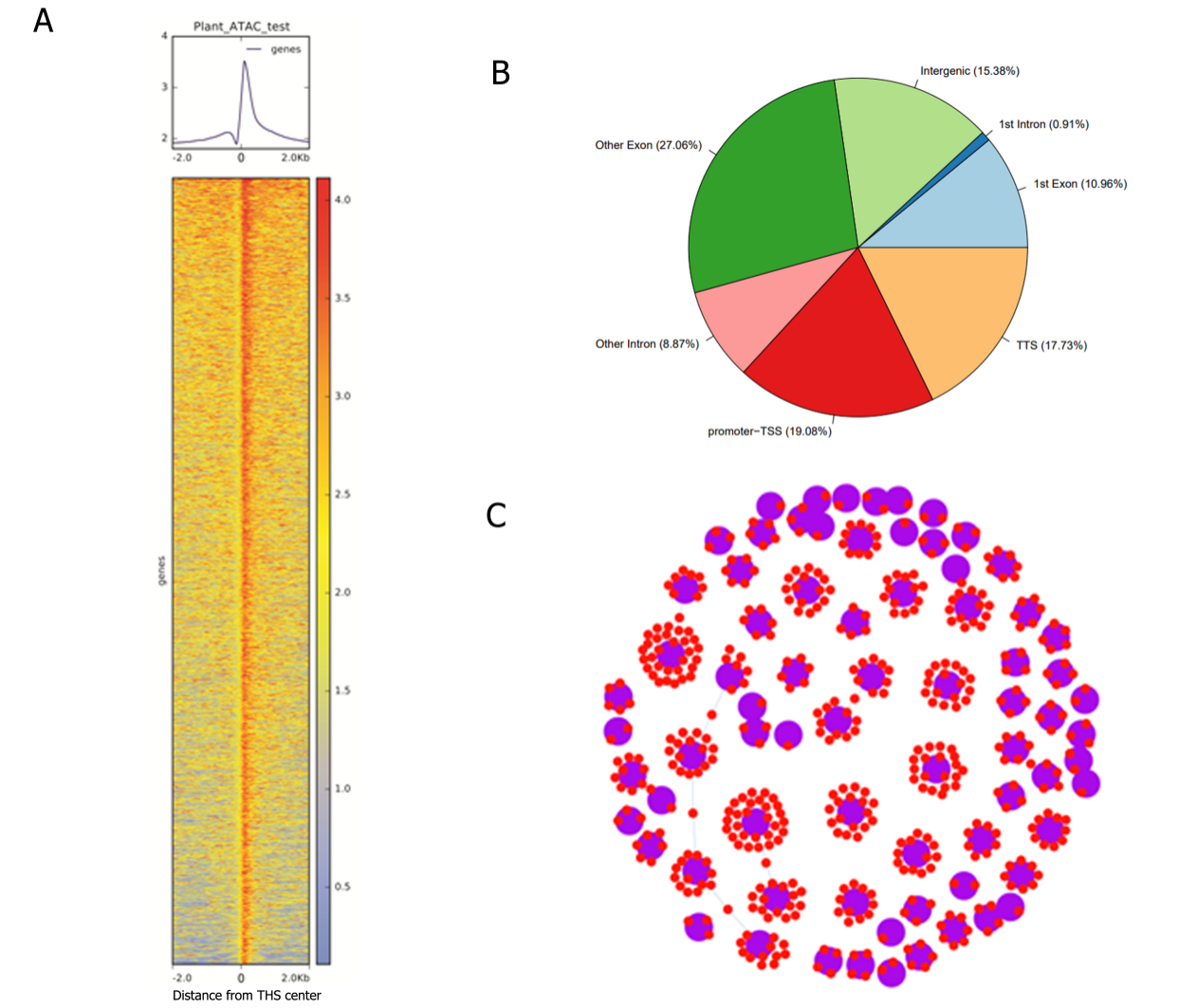
A. 染色质开放区域THSs热图展示
B. peak分布区域展示
C.转录因子靶基因预测,紫色代表转录因子或转录因子家族,红色代表TF可能通过motif作用的靶基因

